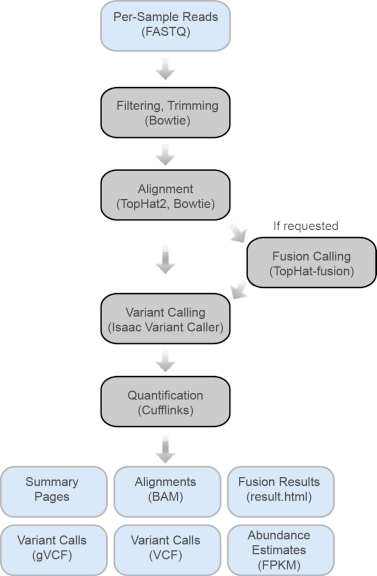
TopHat App Workflow
| 1 | Filtering. The first step of the workflow is to filter the input reads against abundant sequences, such as mitochondrial or ribosomal sequences, as defined by iGenomes (support.illumina.com/sequencing/sequencing_software/igenome.ilmn). Also. The workflow has an internal library of such sequences that it uses for this purpose, including mitochondrial and ribosomal sequences. Bowtie (see Bowtie) performs this alignment. Only sequences that do not align against abundant sequences are passed through to the next phase of the analysis. Read pairs are filtered if at least one read aligns to an abundant sequence. This filtering step also performs trimming of 2 bases from the 5’ end of the read. This is due to a consistent observation of a high mismatch rate from these two bases in RNA-seq libraries. |
| 2 | Alignment. TopHat2 (see TopHat), using the Bowtie 1 aligner, performs a spliced alignment of the filtered reads against the genome. Based on the user-specified genome, TopHat uses a list of known transcripts to align reads against known transcripts and splice junctions. |
| 3 | Fusion Calling: If requested, Tophat-fusion detects gene fusions. Fusion calling occurs in two steps. First, TopHat2 is run in a mode to allow the detection of fused alignments. Then, a post-alignment analysis script identifies candidate fusion genes from these fusion alignments. This step does not work with STAR alignments. |
| 4 | Variant Calling: The Isaac Variant Caller (see Isaac Variant Caller) performs variant calling, producing gVCF output. For stranded library preps, the strand bias filter will be disabled. In addition, the workflow makes use of the -bsnp-diploid-het-bias parameter to expand the allowable range for the heterozygous variant call, in order to account for allele-specific expression. |
| 5 | Quantification: Cufflinks (see Cufflinks) is used for quantification of reference genes and transcripts. |
Figure 9 TopHat App Workflow