FASTQ 파일 설명
10/26/21
Illumina 시퀀싱 기술은 클러스터 생성 및 합성에 의한 시퀀싱(SBS) 화학을 사용하여 시퀀싱 플랫폼에 따라 플로우 셀에서 수백만 또는 수십억 개의 클러스터를 시퀀싱합니다. SBS 화학 검사 중에 각 클러스터에 대해 기기의 실시간 분석(RTA) 소프트웨어에 의한 시퀀싱의 모든 사이클에 대해 기본 호출이 수행되고 저장됩니다. RTA는 기본 통화 데이터를 개별 기본 통화(또는 BCL) 파일 형식으로 저장합니다. 시퀀싱이 완료되면 BCL 파일의 기본 호출을 시퀀스 데이터로 변환해야 합니다. 이 과정을 BCL에서 FASTQ로의 전환이라고 합니다.
FASTQ 파일은 플로우 셀에서 필터를 통과하는 클러스터의 시퀀스 데이터를 포함하는 텍스트 파일입니다(필터를 통과하는 클러스터에 대한 자세한 내용은 이 게시판의 “추가 정보” 섹션을 참조). 샘플이 멀티플렉싱된 경우 FASTQ 파일 생성의 첫 번째 단계는 디멀티플렉싱입니다. 디멀티플렉싱은 클러스터의 인덱스 시퀀스(들)를 기반으로 클러스터를 샘플에 할당합니다. 디멀티플렉싱 후, 조립된 시퀀스는 샘플당 FASTQ 파일에 기록됩니다. 샘플이 멀티플렉싱되지 않은 경우 디멀티플렉싱 단계가 발생하지 않으며 각 플로우 셀 레인에 대해 모든 클러스터가 단일 샘플에 할당됩니다.
단일 리드 런의 경우 플로우 셀 레인당 각 샘플에 대해 하나의 리드 1(R1) FASTQ 파일이 생성됩니다. 페어드 엔드 런의 경우 각 레인의 각 샘플에 대해 하나의 R1 및 하나의 Read 2(R2) FASTQ 파일이 생성됩니다. FASTQ 파일은 *.fastq.gz 확장자로 압축 및 생성됩니다.
FASTQ 파일은 어떤 모습인가요?
필터를 통과하는 각 클러스터에 대해 단일 시퀀스가 해당 샘플의 R1 FASTQ 파일에 기록되고 페어드 엔드 런의 경우 단일 시퀀스가 샘플의 R2 FASTQ 파일에도 기록됩니다. FASTQ 파일의 각 항목은 4줄로 구성됩니다.
- 시퀀싱 런 및 클러스터에 대한 정보가 포함된 시퀀스 식별자. 이 라인의 정확한 내용은 사용된 BCL에서 FASTQ로의 변환 소프트웨어에 따라 다릅니다.
- 시퀀스(기본 호출, A, C, T, G 및 N).
- 단순히 더하기(+) 기호인 구분 기호.
- 기본 통화 품질 점수 . 이는 숫자 품질 점수를 나타내는 ASCII 문자를 사용하여 Phred +33 인코딩됩니다.
다음은 R1 FASTQ 파일의 단일 항목의 예입니다.
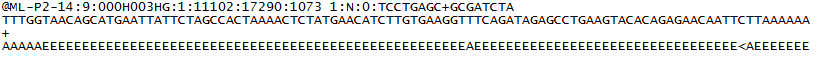
FASTQ 시퀀스 파일 형식에 대한 자세한 내용은 여기에서 확인할 수 있습니다.
FASTQ 파일을 보는 방법
FASTQ 파일에는 최대 수백만 개의 항목이 포함될 수 있으며 크기가 몇 메가바이트 또는 기가바이트일 수 있으므로 보통 텍스트 편집기에서 열기에는 너무 큽니다. 일반적으로 FASTQ 파일은 참조 또는 de novo 어셈블리에 대한 정렬과 같이 다운스트림 분석을 수행하는 도구의 입력으로 사용되는 중간 출력 파일이므로 볼 필요가 없습니다.
문제 해결을 위해 또는 호기심을 벗어나 FASTQ 파일을 확인해야 하는 경우, 매우 큰 파일을 처리할 수 있는 텍스트 편집기 또는 명령줄을 통해 큰 파일을 볼 수 있는 Unix 또는 Linux 시스템에 대한 액세스가 필요합니다.
FASTQ 파일 생성 방법
FASTQ 파일 생성은 MiSeq Reporter가 MiSeq에서 사용하고 Local Run Manager가 MiniSeq에서 사용하는 모든 분석 워크플로우의 첫 단계입니다. 분석이 완료되면 FASTQ 파일은 MiSeq의 <run folder>\Data\Intensities\BaseCalls 및 MiniSeq의 <output folder>\Alignment_#\<subfolder>\Fastq에 위치합니다.
BaseSpace Sequence Hub에 업로드된 모든 런의 경우, 런이 완전히 업로드된 후 FASTQ 파일 생성이 자동으로 발생하며, FASTQ 파일은 BaseSpace Sequence Hub 의 다양한 분석 앱에 대한 입력으로 사용됩니다. BaseSpace Sequence Hub()에서 런과 관련된 프로젝트에서 FASTQ 파일을 찾을 수 있습니다.
bcl2fastq 변환 소프트웨어는 모든 현재 Illumina 시퀀싱 시스템에서 생성된 데이터로부터 FASTQ 파일을 생성하는 데 사용할 수 있습니다.
FASTQ 파일 생성 중에 적용할 수 있는 다양한 설정에 대한 자세한 내용은 아래 소프트웨어 사용 설명서를 참조하십시오.
추가 정보
- 클러스터가 필터를 통과하기 위한 설명 및 요구 사항은 MiSeq의 섹션 1.5.8에서 확인할 수 있습니다. 이미징 및 Base Calling 온라인 교육 과정.
- NovaSeq, NextSeq 500/550 및 MiniSeq 시스템의 기본 호출에 대한 자세한 내용은 2-Channel SBS 기술을 참조하십시오.
- MiSeq 및 HiSeq 시스템 기반 호출에 대한 자세한 내용은 Illumina 시퀀싱 기술을 참조하십시오.