Population Mode
DRAGEN provides a population-based analysis option to jointly analyze samples from unrelated individuals. To initiate population mode, use the following genotypers.
| • | gVCF Genotyper—Uses a set of single or multisample gVCFs as input and outputs a multisample VCF, which contains one entry for any variant seen in any of the input gVCFs. The variants are genotyped across all input samples using information from the hom-ref blocks as necessary. The gVCF genotyper does not adjust genotypes based on population information. See gVCF Genotyper Options for information on the available command line options. |
| • | Joint Genotyper—Uses information from the whole cohort to improve the accuracy of individual genotypes. You can input multisample VCF, multisample gVCF, or a set of single sample gVCFs. To receive output as a multisample gVCF, set --enable-multi-sample-gVCF to true. See Joint Genotyper Options for information on the available command line options. |
| • | Combine gVCFs—Uses single-sample gVCF and multisample gVCFs to generate one multisample gVCF. Variants present in any input sample are genotyped across all samples similarly to gVCF genotyper. Combine gVCF accepts any combination of single-sample gVCFs and multisample gVCFs as input, but generating output might be slow if merging a large number of samples. For large scale population calling, use gVCF Genotyper instead of combine gVCF. See Combine gVCF Options for information on the available command line options. |
The following figure displays the different pathways and data flows between the gVCF Genotyper, Combine gVCFs, and Joint Genotyper.
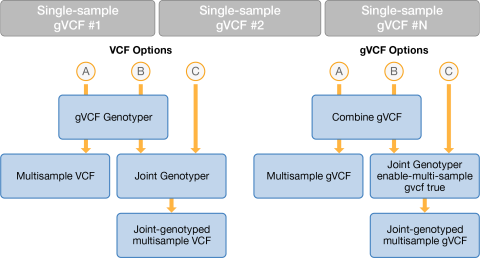
To receive a list of variants present in the cohort and the genotypes of that variant in each of the cohort members, run the gVCF Genotyper. Optionally, you can run the Joint Genotyper after to build a second multisample VCF. The Joint Genotyper output refines the sample genotypes based on the population information. If using the gVCF Genotyper output only, you can filter out infrequent variants to prevent noise if the variants contain low depth or genotype quality. Use an open-source utility like bcftools on the output file to filter the variants.
To compare multiple pedigrees, you can run gVCF Genotyper on the output of Joint Genotyper and merge multiple joint-called pedigrees into a single multisample VCF.
Use the --vc-emit-ref-confidence gVCF option to configure the Joint Genotyper to write a multisample GVCF.