Somatic CNV Calling
To detect somatic copy number aberrations and regions with loss of heterozygosity, run the DRAGEN CNV caller on a tumor sample in combination with a VCF containing germline SNVs. The output file is a VCF file. Components of the germline CNV caller are reused in the somatic algorithm with the addition of a somatic modeling component, which estimates tumor purity and ploidy.
Somatic CNV Caller Workflow
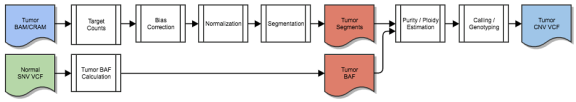
The germline SNVs are used to compute b-allele ratios in the tumor, which allows for allele-specific copy number calling on the tumor sample. Where possible, use of the small-variant VCF from a matched normal sample is preferred ("tumor/normal" mode), but a catalog of population SNPs can be used when a matched normal sample is not available ("tumor-only" mode).
When a matched normal sample is available, it should first be processed using the germline small variant caller. In this case, only germline-heterozygous SNV sites are used for determining b-allele ratios. If no matched normal is available, population SNP b-allele ratios are computed as for matched normal heterozygous loci, but are treated as variants of unknown germline genotype; possible genotype assignments are statistically integrated to determine allele specific copy number.
In matched normal mode, a VCF containing germline copy number changes for the individual may optionally be input. This ensures that germline CNVs are output as separate segments in the somatic CNV VCF, and annotated with the germline copy number so that it is clear whether there are specifically-somatic copy number changes in the region.