Introduction
Genetic diseases are a leading cause of pediatric deaths, particularly in infants admitted to neonatal intensive care units (NICUs). 1,2 Approximately 40% of neonatal deaths are associated with a rare genetic disease.1-5 Due to extremely rapid disease progression in pediatric patients with genetic disease, it is crucial that the causative genetic variation is quickly identified. Rapid diagnosis can allow for timely application of therapeutic interventions, preventing severe morbidity and mortality in this vulnerable patient population.6 Time to diagnosis using standard genome sequencing techniques may take up to several weeks and requires interpretation and management decisions by a healthcare provider. These delays in diagnosis and initiation of treatment may result in poorer outcomes in some acutely ill patients with rare genetic diseases.
Rapid whole-genome sequencing (rWGS) is an alternative to standard genome sequencing, with the potential to offer fast and accurate diagnosis of rare genetic disease in critically ill infants in under 50 hours.7 In clinical studies, rWGS has demonstrated a positive diagnosis rate in up to 57% of cases,8 and informed medical management in 30% to 72% of cases in pediatric patients admitted to the ICU.7-13 However, the clinical utility of rWGS has been limited by lack of scalability and the inability to resolve complex structural variation that contributes to 20% of childhood genetic diagnoses in the ICU.14
In this article, we describe our involvement in researching and developing a new, reengineered genome sequencing workflow for improved speed, reproducibility, and scalability, providing rWGS is 13.5 hours. This pipeline, developed in collaboration with Rady Children’s Institute for Genomic Medicine, and powered by Illumina next-generation sequencing (NGS) technology, eliminates critical bottlenecks in conventional rWGS workflows, by optimizing library preparation, sequencing, and variant calling, to provide fast and accurate identification of genetic variants, including complex structural variation.
Optimized 13.5-hour genome sequencing workflow
From previous work we reported a rWGS pipeline that correctly identified causative genetic variation in under 20 hours.9 Though promising, this methodology lacked scalability and did not adequately capture structural variation. In this report, we describe further innovations to optimize the rWGS workflow, improving scalability, turnaround time, and analytic performance for copy number variants (CNVs) and structural variants (SVs) (Figure 1).

VC, variant calling; SV, structural variant; TSS, TruSight Software Suite; GEM, Fabric GEM genomic interpretation platform; MOON, Invitae variant interpretation software. *660 minutes was using a tailored run recipe and maximally reduced cycle times.
Accelerated library preparation with Illumina DNA PCR-Free Prep
A key challenge in sample preparation for rWGS is the purification of high-quality genomic DNA from low sample volumes. We prepared sequencing libraries directly from ethylenediaminetetraacetic acid (EDTA) blood samples or five 3-mm punches from a Nucleic Card Matrix dried blood spot (Thermo Fisher, Catalog no. 4473977), which are the sample types frequently used in mandatory newborn screening. Illumina DNA PCR-Free Prep, Tagmentation, which uses bead-linked transposomes to deliver consistent insert sizes while reducing hands-on time, was used for library preparation. On-bead tagmentation eliminates the need for intermediate DNA purification steps while retaining the ability to identify CNVs and SVs. Incubation times were maximally shortened to normalize library output. This allowed faster measurement of library concentration using the Library Quantification Kit (KAPA Biosystems, Catalog no. KK4824-079601666001). Overall, library preparation was completed in an average of 45 minutes from purified genomic DNA, and 72 minutes from blood.
High-performance sequencing using the NovaSeq™ 6000 System
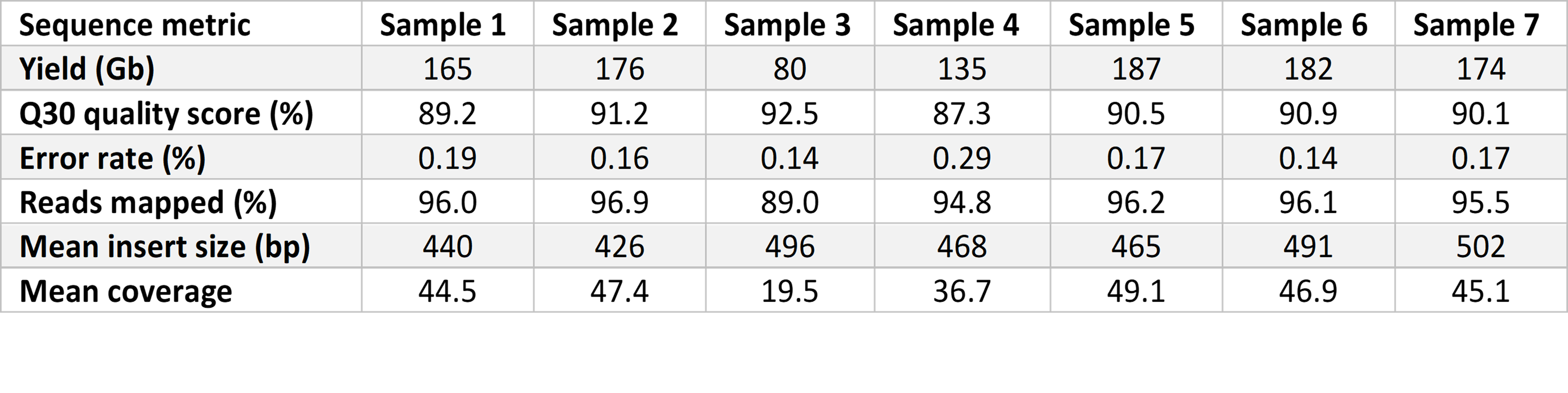
Prepared libraries were sequenced on the NovaSeq 6000 System with a 2 × 101 bp cycle using SP flow cells with version 1.5 reagents for high-quality sequencing output. Flow cells were imaged on a single surface of each of two lanes. We used a tailored run recipe with maximally reduced cycle time, allowing sequence quality to be retained. Sequencing yield was approximately 150 Gb at over 40× coverage and 87% reads with Phred quality scores over 30 (Q30) (Table 1). The average time for sequencing runs was 11 hours 12 minutes.
Faster sequence alignment and variant calling with Dynamic Read Analysis for GENomics (DRAGEN™ v.3.7)
The Illumina DRAGEN platform, which is highly optimized for speed, sensitivity, and accuracy, was used for sequence alignment and variant calling. Transfer of files from the NovaSeq 6000 sequencing system to the DRAGEN platform was automated. Sequences were aligned to human genome assembly GRCh37 (hg19) and variants were identified and genotyped with the DRAGEN platform v.3.7.5. Variant call files were automatically routed to variant interpretation software, including Illumina TruSight™ Software Suite and third-party software, MOON (Invitae) and GEM (Fabric Genomics). Tertiary analysis was run in parallel on all three software platforms. Sequential filtering and ranking variants using decision trees, Bayesian models, neural networks, and natural language processing, were applied to generate outputs for variant identification.
The analytical performance of this improved rWGS pipeline was assessed using two reference DNA samples (NA12878 and NA24385). These reference samples contain gold standard variant sets established by the National Institute of Standards and Technology (NIST) for SNVs and insertion-deletions (indels) (NISTv4.1) and SVs and CNVs (NISTv0.6). Witty.Er was used to calculate analytical performance for CNV and SV detection. Average time from DNA sample preparation to completion of variant calling was 12 hours 42 minutes, a 35% improvement compared to the previous benchmark using DRAGEN v3.5.3 (Figure 2).
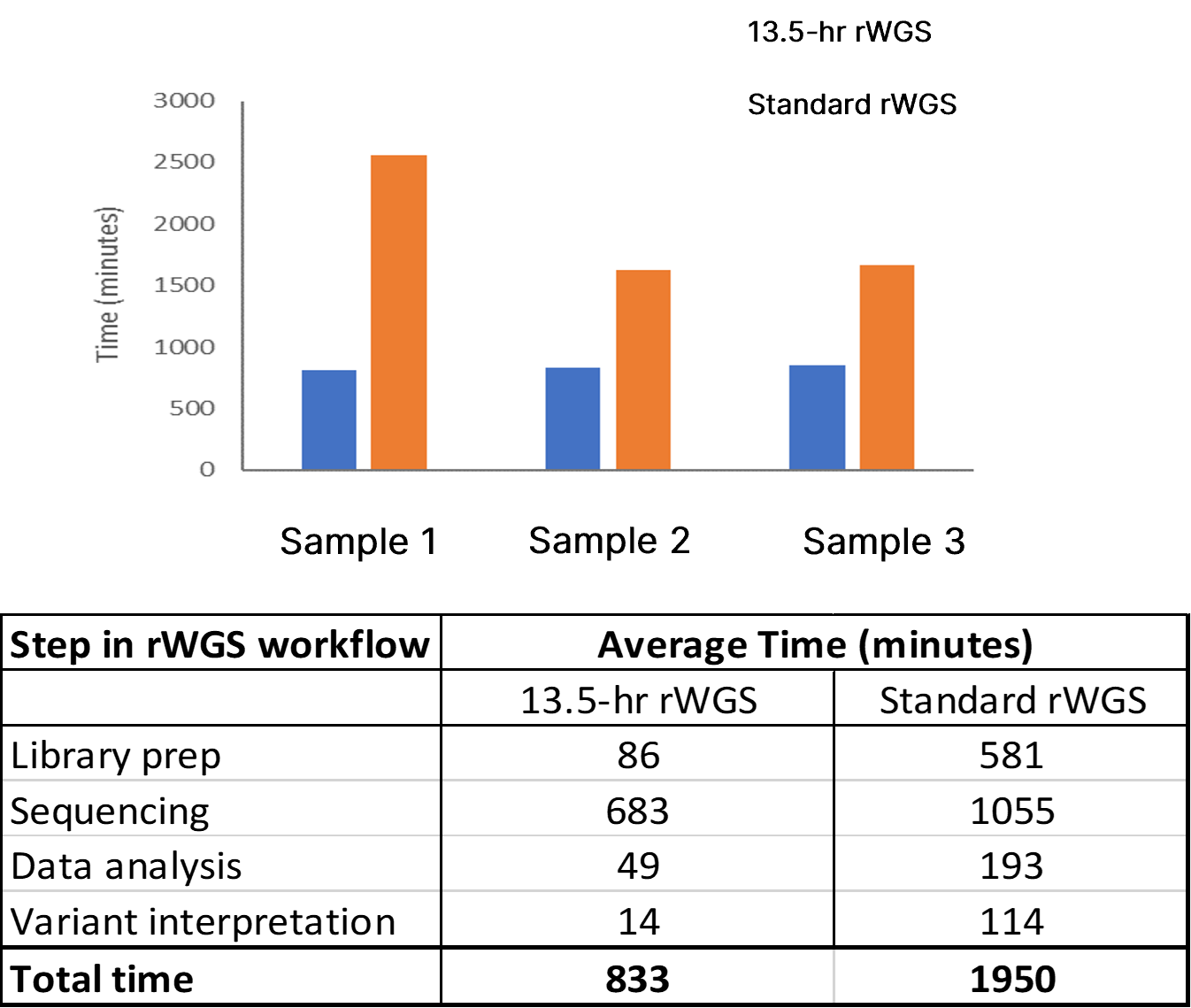
Top panel: Total time to completion in three test samples using 13-hr rWGS (blue) and standard rWGS (orange) methods. Bottom panel: Average time taken at each step of the rWGS workflow using the 13.5-hr and standard rWGS pipelines.
Superior analytic performance of DRAGEN v.3.7 for structural variant detection
The performance of DRAGEN v.3.7 was compared with Manta and CNVnator, which are commonly used tools for detection of SVs (size > 50 nt) and CNVs (size > 10 kb). DRAGEN v.3.7 demonstrated superior sensitivity for insertion SVs (49% with DRAGEN vs 27% with Manta) and deletion CNVs (88% with DRAGEN vs 9% with CNVnator) (Table 2). Average time for primary and secondary analysis, including conversion of raw data from base call to FASTQ format, alignment to reference genome, and variant calling, was significantly improved at 47 minutes using DRAGEN v.3.7, compared to 3 hours 9 minutes using Manta and CNVnator.

Precision and recall using DRAGEN v.3.7 and Manta/CNVnator in reference sample NA24385, using NIST benchmarks were significantly improved for detection of CNV deletion. SNV, single nucleotide variation; indel, insertion/deletion; SV, structural variant (size > 50 bp); CNV, copy number variation (size > 10 kb).
Potential utility of the 13.5-hr rWGS pipeline
The 13.5-hr rWGS pipeline provided correct identification of genetic variation in six out of seven test samples. Causative genetic variation was identified in all samples in under 14 hours using our rWGS workflow compared to an average of 42 hours by standard rWGS. The fastest time to completion was 13 hours 13 minutes. Our rWGS workflow combines speed with reproducibility and scalability, making it well suited for generalizable, high-throughput applications.
Summary
In this article we outline a reengineered rWGS pipeline that has been optimized to provide fast and accurate identification of causative genetic variation in test samples. Key steps in the rWGS workflow, including accelerated library preparation and high-performance sequencing with the NovaSeq 6000 System, combined with the precision and speed of DRAGEN v.3.7 variant calling, allowed rWGS to be completed in 13.5 hours. In addition to the improvements in sequencing speeds and compatibility with third-part variant interpretation tools, this rWGS pipeline enables researchers to rapidly detect variants, including structural variation, with high accuracy.
Read full publication: https://www.nature.com/articles/s41467-022-31446-6
References
1. Gunne E, McGarvey C, Hamilton K, Treacy E, Lambert DM, Lynch SA. A retrospective review of the contribution of rare diseases to paediatric mortality in Ireland. Orphanet J Rare Dis. 2020;15(1):311. doi:10.1186/s13023-020-01574-7Weiner
2. Weiner J, Sharma J, Lantos J, Kilbride H. How infants die in the neonatal intensive care unit: trends from 1999 through 2008. Arch Pediatr Adolesc Med. 2011;165(7):630-634. doi:10.1001/archpediatrics.2011.102
3. Murphy SL, Xu J, Kochanek KD, Arias E. Mortality in the United States, 2017. NCHS Data Brief. 2018(328):1-8.
4. Arth AC, Tinker SC, Simeone RM, Ailes EC, Cragan JD, Grosse SD. Inpatient Hospitalization Costs Associated with Birth Defects Among Persons of All Ages - United States, 2013. MMWR Morb Mortal Wkly Rep. 2017;66(2):41-46.
5. Berry MA, Shah PS, Brouillette RT, Hellmann J. Predictors of mortality and length of stay for neonates admitted to children's hospital neonatal intensive care units. J Perinatol. 2008;28(4):297-302.
6. Petrikin JE, Cakici JA, Clark MM, et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom Med. 2018;3:6. doi:10.1038/s41525-018-0045-8
7. Saunders CJ, Miller NA, Soden SE, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4(154):154ra135. doi:10.1126/scitranslmed.3004041
8. Petrikin JE, Willig LK, Smith LD, Kingsmore SF. Rapid whole genome sequencing and precision neonatology. Semin Perinatol. 2015;39(8):623-631. doi:10.1053/j.semperi.2015.09.009Farnaes
9. Clark MM, Hildreth A, Batalov S, et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci Transl Med. 2019;11(489):eaat6177. doi:10.1126/scitranslmed.aat6177
10. Kingsmore SF, Cakici JA, Clark MM, et al. A Randomized, Controlled Trial of the Analytic and Diagnostic Performance of Singleton and Trio, Rapid Genome and Exome Sequencing in Ill Infants. Am J Hum Genet. 2019;105(4):719-733. doi:10.1016/j.ajhg.2019.08.009
11. Farnaes L, Hildreth A, Sweeney NM, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3:10. doi:10.1038/s41525-018-0049-4
12. Willig LK, Petrikin JE, Smith LD, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3(5):377-387. doi:10.1016/S2213-2600(15)00139-3
13. Sanford EF, Clark MM, Farnaes L, et al. Rapid Whole Genome Sequencing Has Clinical Utility in Children in the PICU. Pediatr Crit Care Med. 2019;20(11):1007-1020. doi:10.1097/PCC.0000000000002056
14. Maron JL, Kingsmore SF, Wigby K, et al. Novel Variant Findings and Challenges Associated With the Clinical Integration of Genomic Testing: An Interim Report of the Genomic Medicine for Ill Neonates and Infants (GEMINI) Study. JAMA Pediatr. 2021;175(5):e205906. doi:10.1001/jamapediatrics.2020.5906