시퀀싱 커버리지
NGS에서 커버리지란 무엇인가요?
차세대 시퀀싱(Next-generation sequencing, NGS) 커버리지란 알려진 참조 염기에 정렬(alignment)되거나 이를 “커버하는” 리드의 평균 수를 의미합니다. 시퀀싱 커버리지 수준을 통해 특정 염기 위치에서 어느 정도의 신뢰도로 변이를 발견할 수 있는지 여부가 결정되는 경우가 많습니다.
시퀀싱 커버리지의 요건은 아래 언급된 바와 같이, 애플리케이션에 따라 차이가 있습니다. 커버리지 수준이 높을수록, 각 염기는 더 많은 수의 정렬된 시퀀스 리드 수로 커버되므로, 더 높은 신뢰도로 베이스 콜이 이루어질 수 있습니다.
시퀀싱 커버리지 권장 사항
연구자들은 일반적으로 사용하는 방법뿐만 아니라 참조 유전체(reference genome) 크기, 유전자 발현 수준, 관심을 가지고 있는 특정 애플리케이션, 발표된 논문, 학계의 모범 사례와 같은 다른 요인들에 근거하여 필요한 NGS 커버리지 수준을 결정합니다. 몇 가지 일반적인 방법에 대한 시퀀싱 커버리지 권장 사항의 예시는 이와 같습니다.
| 시퀀싱 방법 | 권장되는 커버리지 |
|---|---|
| 전장 유전체 시퀀싱(whole-genome sequencing, WGS) | 인간 WGS의 경우 30~50x(애플리케이션 및 통계 모델에 따라 차이가 있음) |
| 전장 엑솜 시퀀싱 | 100x |
| RNA 시퀀싱 | 일반적으로 샘플링되는 리드 수(단위: M)를 기준으로 계산됨. 드물게 발현되는 유전자 검출은 커버리지 뎁스를 높여야 하는 경우가 많음. |
| ChIP-Seq | 100x |

원하는 NGS 커버리지 수준을 추정하고 달성하는 방법
시퀀싱 런 예상:
Lander/Waterman 공식1은 유전체 커버리지를 산출하는데 사용되는 공식입니다. 일반적인 공식: C = LN / G
- C: 커버리지
- G: 반수체 유전체 길이
- L: 리드 길이(read length)
- N: 리드 수
Illumina는 과학자들이 커버리지를 결정하는 데 도움이 되는 다음과 같은 리소스를 제공합니다.
- 시퀀싱 커버리지 계산기: 실험 수행 시 원하는 시퀀싱 커버리지를 달성하는 데 필요한 시약 및 시퀀싱 런을 계산하는 방법을 확인하실 수 있습니다.
- RNA-Seq 리드 길이 및 커버리지: 여러 건의 RNA-Seq 프로젝트에 적합한 시퀀싱 리드 뎁스에 관한 가이드라인을 확인하실 수 있습니다.
추가 시퀀스 분석이 필요한 경우
추가 자료가 필요한 경우 커버리지 또는 시퀀스 뎁스를 늘릴 수 있습니다. 필요 시 서로 다른 플로우 셀의 시퀀싱 아웃풋을 원본 샘플과 통합할 수 있습니다. 다음과 같은 이유로 기존 예상 커버리지보다 더 많은 시퀀스를 분석해야 할 수 있습니다.
- Assay의 통계 검정력을 증가시키기 위해
- 매우 드문 사례를 조사하는 경우
- 특정 학술지 또는 연구 분야의 커버리지 최소 한도를 충족하기 위해
- 시퀀스를 분석하기 어려운 영역 또는 다배수체 유전체의 시퀀스를 분석하는 경우
NGS 커버리지 영역 및 균일성을 나타내는 히스토그램
커버리지 히스토그램은 일반적으로 전체 데이터 세트의 시퀀싱 커버리지 범위와 균일성을 나타내는 데 사용됩니다. 이는 다양한 뎁스에서 매핑된 시퀀싱 리드로 커버되는 참조 염기의 수를 나타내어 전체적인 커버리지 분포를 보여줍니다. 매핑된 리드 뎁스는 주어진 참조 염기 위치에 시퀀스 분석 및 정렬된 염기의 총 수를 나타냅니다(시퀀싱 분야의 경우 “매핑”과 “정렬”은 같은 의미로 사용됨).
시퀀싱 커버리지 히스토그램에서 리드 뎁스는 구간별로 분류하여 X축에 표시하고, 각 리드 뎁스 구간별 참조 염기의 총 개수를 Y축에 표시합니다. 참조 염기의 백분율 또한 표시할 수 있습니다.
커버리지 히스토그램의 예시
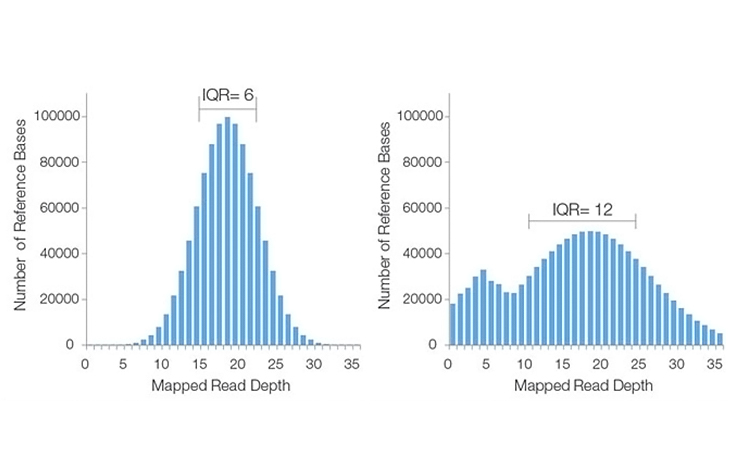
이상적으로는, 이 히스토그램 이미지와 같이, 표준 편차가 작은 포아송 유사 분포 형태를 취하게 됩니다. 이 분포는 리드가 유전체 전체에 무작위로 분포되어 있고 1회의 시퀀싱 런 내에서 리드 간 실제 중복(overlap)을 검출하는 능력이 일정하다는 가정 하에 유효합니다.
그러나 오른쪽의 시퀀싱 커버리지 히스토그램의 바람직하지 않은 예시와 같이, 여러 가지 이유로 인해, 실제 커버리지 히스토그램은 넓게 퍼져 있을 수 있거나(즉, 리드 뎁스의 폭이 넓음), 비포아송 분포를 가질 수 있습니다.
차세대 시퀀싱 커버리지의 평가
NGS 커버리지를 평가하는 데 흔히 사용되는 지표는 다음과 같습니다.
사분위수 범위(Inter-Quartile Range, IQR)
IQR이란 히스토그램의 75번째 백분위수와 25번째 백분위수의 시퀀싱 커버리지 차이를 뜻합니다. 이 값은 통계적 변동성을 측정하는 값으로, 데이터 세트 전체에 걸친 커버리지의 불균일성을 나타냅니다.
IQR이 높다는 것은 유전체 전체에 걸쳐서 커버리지의 편차가 크다는 것을 나타내는 반면, IQR이 낮다는 것은 시퀀스 커버리지의 균일성이 높다는 것을 나타냅니다. 위 히스토그램 예시에서, 왼쪽 히스토그램의 IQR이 더 낮다는 것은 오른쪽 히스토그램보다 시퀀싱 커버리지 균일성이 더 높다는 것을 나타냅니다.
평균 (매핑된) 리드 뎁스
평균 매핑된 리드 뎁스(또는 평균 리드 뎁스)는 각 참조 염기 위치에 매핑된 리드 뎁스의 합계를 참조 유전체의 알려진 염기의 수로 나눈 값입니다.
평균 리드 뎁스 지표는 얼마나 많은 수의 리드가 주어진 참조 염기 위치에 정렬될 가능성이 있는지를 나타냅니다.
미가공 리드 뎁스
이는 기기가 생성한 시퀀스 데이터의 총량(정렬[alignment] 전)을 참조 유전체 크기로 나눈 값입니다. 시퀀싱 기기 공급업체에서 미가공 리드 뎁스를 사양에 표시했더라도, 이는 정렬 과정의 효율성을 고려하지 않은 값입니다.
정렬 과정 중에 raw 시퀀싱 리드의 상당 부분이 폐기되는 경우, 정렬 후 매핑된 리드 뎁스가 미가공 리드 뎁스에 비해 훨씬 작을 수 있습니다.
Interested in receiving newsletters, case studies, and information from Illumina based on your area of interest? Sign up now.
참고문헌
- Lander ES, Waterman MS. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics. 1988;2(3):231-239. doi:10.1016/0888-7543(88)90007-9